
La notación D y L para la configuración absoluta de azúcares y aminoácidos
¿Qué diferencia «D – glucosa» de «l-glucosa» ? ¿O D-alanina de L-alanina?
¿y qué es esta nomenclatura D – y L -, de todos modos? Sigue leyendo:
tabla de contenidos
- D – y L – proporciona una abreviatura rápida para designar enantiómeros
- ¿Por qué nos molestamos con esta Nomenclatura Antigua?,
- El sistema L-Y D-para asignar «configuración absoluta»
- aldehído de cuatro carbonos D-y L-azúcares (Aldotetroses)
- aldehído de cinco carbonos D-y L-azúcares (Aldopentosis)
- aldehído de seis carbonos D-y L – azúcares (Aldohexosis)
- Pero espera – ¡hay más! (Aminoácidos)
- Resumen: Notación D – y L – para azúcares y aminoácidos
- notas
- referencias (avanzadas) y lectura adicional
D – y L – proporciona una abreviatura rápida para designar enantiómeros
la notación D – y L – proporciona una abreviatura rápida para designar enantiómeros.,
La D-glucosa es el enantiómero de la L-glucosa, por ejemplo. Como L-alanina es el enantiómero de D-alanina.
se asigna de la siguiente manera. Para un azúcar dibujado en la proyección de Fischer con el carbono más oxidado en la parte superior (es decir, aldehído o cetona)
- si el OH en el centro quiral inferior apunta hacia la derecha, se denomina d –
- Si el OH en el centro quiral inferior apunta hacia la izquierda, se denomina L- .
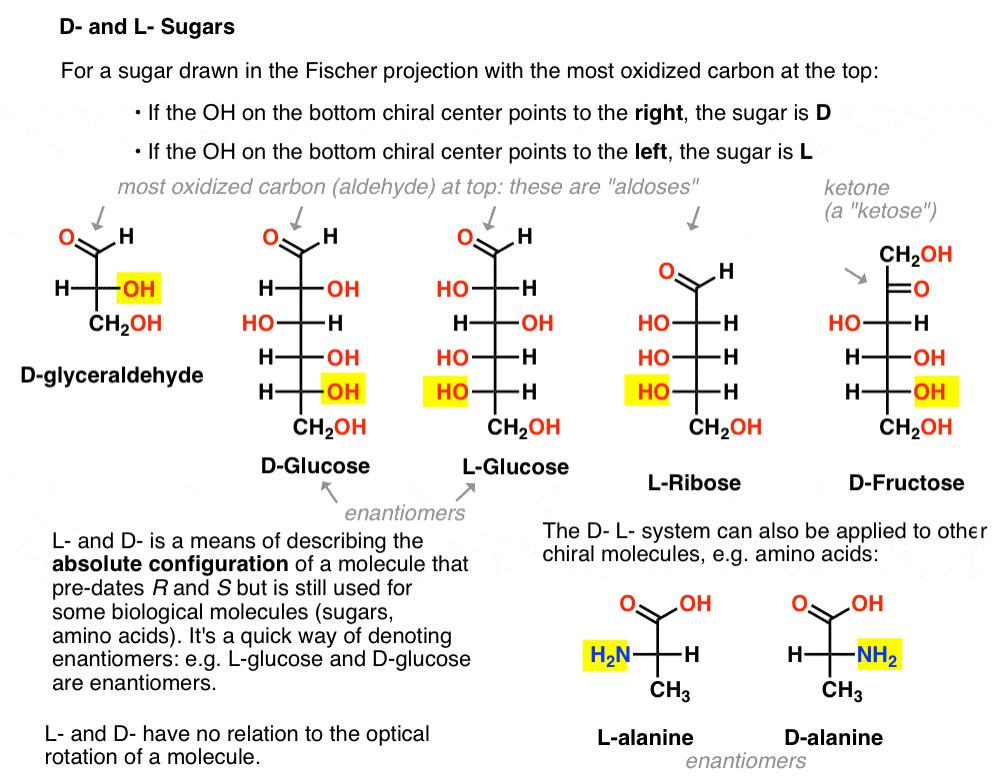
Esta terminología también se puede aplicar a los aminoácidos: véase L – y D – alanina en la imagen de arriba.,
2. ¿Por Qué Nos Molestamos Con Esta Antigua Nomenclatura?
usted podría preguntar justificadamente: ¿no tenemos ya un sistema para asignar la configuración absoluta ? ¿Por qué necesitamos un nuevo sistema?
el sistema D-L no es un sistema nuevo, amigos. Es el viejo sistema-es anterior a Cahn-Ingold-Prelog.
el sistema D-L es literalmente un remanente de la era del caballo y el buggy, que se remonta al trabajo de Emil Fischer sobre los carbohidratos a finales de 1800, una época en que los químicos orgánicos no tenían forma de determinar la configuración absoluta de los estereocentros, lo que solo fue posible en 1951 (thx, Bijvoet).,
entonces, ¿por qué todavía se usa? ¿No debería ser consignado al basurero de la historia, junto con reglas de cálculo, casetes de 8 pistas y disquetes de 5¼»?
Bueno, hay comunidades prósperas en partes de la América rural donde persisten los carruajes tirados por caballos, si sabe dónde buscar. (Tal vez algún día habrá comunas donde la gente solo utiliza la tecnología informática de los años 1970 y 1980?)
del mismo modo hay un bolsillo de química orgánica donde el sistema D-L todavía encuentra uso, y que es específicamente en el Reino de los azúcares y aminoácidos.,
esto no es una revuelta de los químicos Amish contra los males modernos del sistema CIP, por cierto. Hay al menos 3 buenas razones, en el caso específico de azúcares y aminoácidos, para usar L – y D- :
- brevedad. D-glucosa es un infierno de mucho más rápido para escribir y decir que (2R,3S,4R, 5R)2,3,4,5,6-pentahydroxyhexanal. El sistema L-/D permite la configuración de una molécula con múltiples centros quirales para ser resumido con una sola letra (más su nombre común, por supuesto – gracias a Noel para el recordatorio)
- Más brevedad., Resulta ser una forma rápida de referirse a los enantiómeros. El enantiómero de la L-glucosa es la d-glucosa. El enantiómero de L-triptófano es D-triptófano. Y si bien podríamos usar los prefijos ( + ) – o ( – ) – para diferenciar los dos enantiómeros de glucosa y otros azúcares, el signo de rotación óptica puede variar con el disolvente, la temperatura, la concentración y otros factores, lo que lo hace menos que ideal. Además, L – Y D-se refieren específicamente a la configuración absoluta, mientras que (como señalamos anteriormente) no hay una relación simple entre el signo de rotación óptica y la configuración.,
- resulta que la mayoría de los azúcares naturales son D -, y la mayoría de los aminoácidos naturales son L -. Hay una enorme cantidad de información comprimida en esa declaración, y no hay ningún sistema competidor (R/S, +/–) que pueda reemplazar el L – Y D – con un solo carácter. Nota
vale la pena repetir: con azúcares y aminoácidos, L-y D-pueden ser designaciones útiles. Para otras moléculas, puede olvidarse en gran medida de ello.
entonces, ¿qué es este sistema D-/L, y cómo se relacionan estos términos con la estructura?,
Únase a mí mientras viajamos hacia atrás en el tiempo
el sistema L Y D para asignar «configuración absoluta»
Emil Fischer comenzó a estudiar los carbohidratos a finales de la década de 1880. en ese momento se sabía (a través de Van’t Hoff) que el carbono era tetraédrico, y también se sabía que las moléculas que contenían un carbono con cuatro sustituyentes diferentes podían rotar la luz polarizada plana (por ejemplo, Pasteur). Lo que no se conocía era la configuración absoluta de cualquiera de las moléculas quirales – lo que nos referiríamos hoy como sus configuraciones «R» y «S».,
el carbohidrato más simple que contiene un centro quiral es el gliceraldehído, C3H6O3. El gliceraldehído tiene tres carbonos; por lo que es una «triosa». El carbono más oxidado en el gliceraldehído es un aldehído, que también lo convierte en una»Aldosa».

En 1888, los dos enantiómeros del gliceraldehído habían sido aislados y caracterizados., Pero como no hay una correlación simple entre la configuración de un centro quiral y la dirección en la que gira la luz polarizada plana, Emil Fischer no tenía forma de vincular la rotación óptica de ( + ) y (-) gliceraldehído a la configuración absoluta de los átomos alrededor del centro quiral.
Usando la terminología actual, no tenía forma de saber si ( – ) – gliceraldehído era (R) O (S).
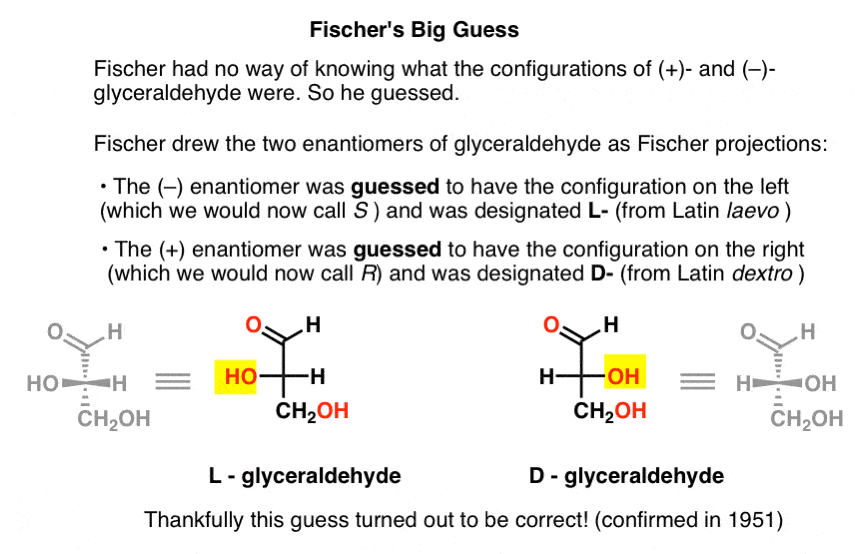
careciendo de esta pieza clave de información, Fischer eligió adivinar.,
la conjetura, que resultó ser correcta, fue que (–)-gliceraldehído tenía la configuración que ahora llamamos S, y que (+)-gliceraldehído tiene la configuración que ahora llamamos R.
Por supuesto, Cahn, Ingold y Prelog aún no habían nacido, y el sistema CIP solo se desarrollaría después del trabajo de Bijvoet en 1951. Así que Fischer desarrolló su propia Nomenclatura.
dibujando gliceraldehído en lo que más tarde se llamaría la proyección de Fischer, asignó la configuración de la izquierda a ( – ) – gliceraldehído, y lo llamó L – (abreviatura del latín laevo )., Luego asignó la configuración de la derecha a (+)-gliceraldehído, y lo llamó D – (para latín dextro).
¿por Qué es esto tan importante?
asignar la configuración absoluta para L-y D-gliceraldehído fue un poco como asignar el meridiano principal (Longitud 0°) al Observatorio Real en Greenwich, Inglaterra., Al igual que la longitud de cualquier otro lugar de la Tierra podría determinarse en relación con ese punto si se conocieran sus distancias relativas, la configuración absoluta de cada otro estereocentro podría determinarse si se conociera su configuración relativa a L – O D – gliceraldehído.
esa podría no ser la analogía más clara. Así que echemos un vistazo a los azúcares de 4 carbonos para otro ejemplo.,
cuatro aldehídos de carbono D – y L – azúcares (Aldotetroses)
Una vez que se propusieron las configuraciones absolutas de l – y D – gliceraldehído, las configuraciones absolutas de otros compuestos quirales podrían establecerse por analogía (y mucho trabajo de grunt químico).
no es crucial para hoy, pero para un ejemplo de este tipo de razonamiento, ver esto .
Cuando se introdujo el concepto de centros quirales, es probable que aprendiera que una molécula con n centros quirales tendrá 2N estereoisómeros (siempre y cuando no haya compuestos meso).,
Hay dos aldosas de cuatro carbonos, treosa y eritrosa. Cada uno tiene dos centros quirales. Cada uno existe como un par de enantiómeros (L – y D- ) dando cuatro estereoisómeros en total.
Aquí están. Lo importante a tener en cuenta en la figura a continuación es que la familia L de azúcares tiene el grupo OH del carbono quiral inferior a la izquierda, y la familia D tiene el grupo OH del carbono quiral inferior a la derecha (resaltado).,
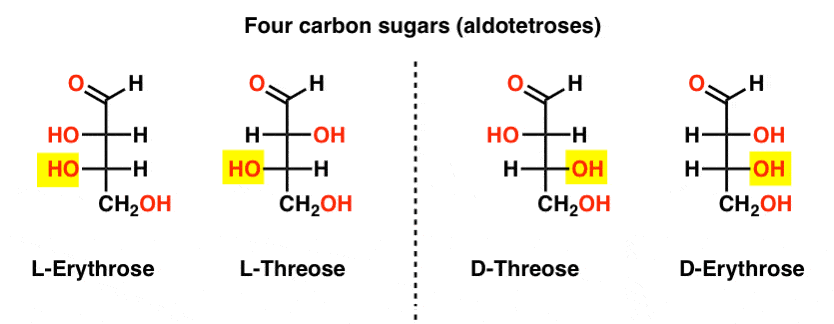
vea cómo L-Erythrose y L-Threose construyen en el estereocentro establecido en L-gliceraldehído (resaltado), y D-Erythrose y D-Threose construyen en el estereocentro establecido en D-gliceraldehído (resaltado).
los azúcares se acumulan un poco como el sistema binario; se puede pensar que cada estereocentro es un » bit » que puede tener uno de dos valores. La configuración de l-erythrose y L-threose difiere solamente en un estereocenter. Si tuviéramos que voltear la posición de H Y OH, tendríamos la otra., Esta relación tiene un nombre que se puede ver a veces: dos moléculas que tienen la configuración opuesta en un solo estereocentro se llaman epímeros, particularmente cuando uno de los átomos unidos al estereocentro es un hidrógeno (H).
cinco aldehídos de carbono D – y L – azúcares (Aldopentosis)
El nombre más familiar en esa lista debe ser ribosa, que es la columna vertebral de azúcar del ácido ribonucleico (ARN).
en el lado izquierdo del diagrama de abajo, tenemos las l-aldopentosis, que comparten la misma configuración del estereocentro inferior cuando el aldehído se coloca en la parte superior.,
sus enantiómeros, las D-aldopentosis, están en el lado derecho, que comparten la misma configuración del estereocentro inferior (resaltado).
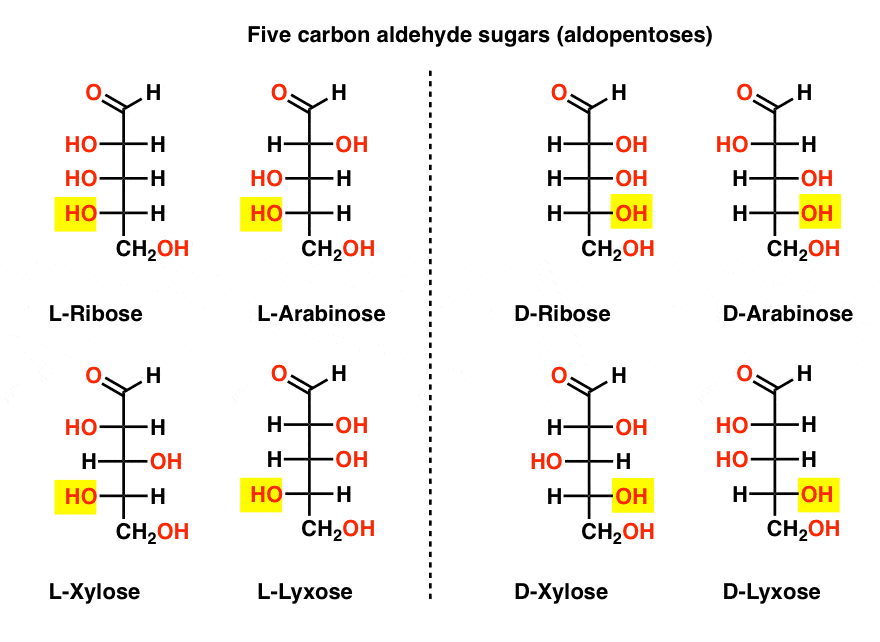
en este punto debemos señalar que la abrumadora mayoría de los azúcares en formas de vida basadas en la Tierra son d-azúcares, incluida la D – ribosa como la columna vertebral del ARN. Por qué y cómo todos los organismos en la tierra terminaron con azúcares D es un misterio, ya que uno supone que los azúcares L habrían funcionado igual de bien.,
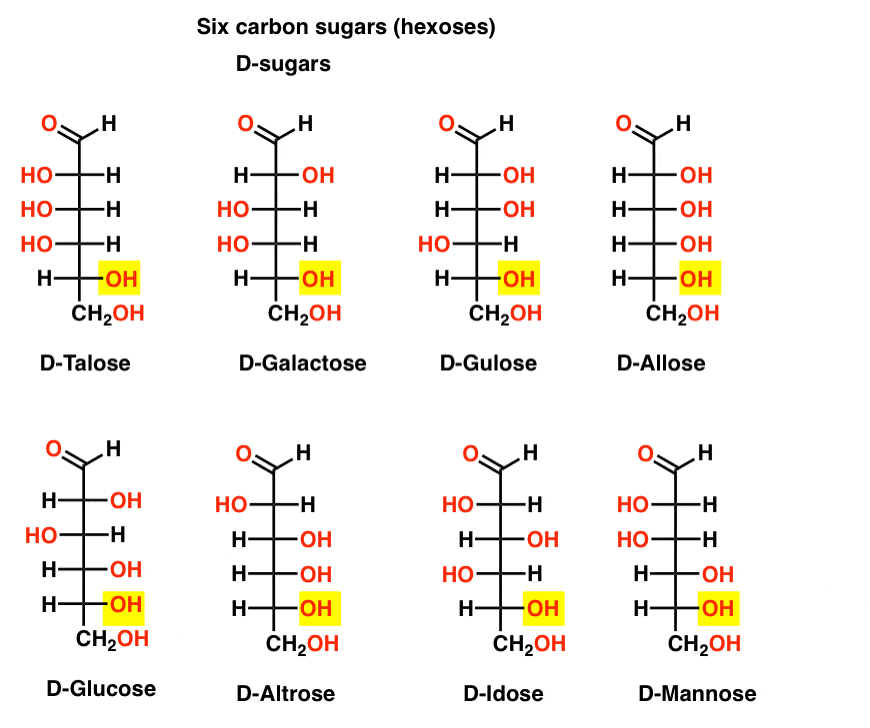
seis aldehídos de carbono D – y L – azúcares (Aldohexosis)
si hay 4 aldopentosis, cada una como un par D – L de enantiómeros (8 estereoisómeros en total), ¿cuántas aldohexosis hay?
Hay 8 pares D-L (16 estereoisómeros en total). La más conocida es la glucosa, pero es probable que reconozcas la manosa y la galactosa. Algunos rara vez, si es que alguna vez, se encuentran en la naturaleza (idose, alguien?).
Aquí están las D-aldohexosis. Observe cómo todos tienen la misma configuración del centro quiral inferior, el mismo que vimos en D-gliceraldehído.,
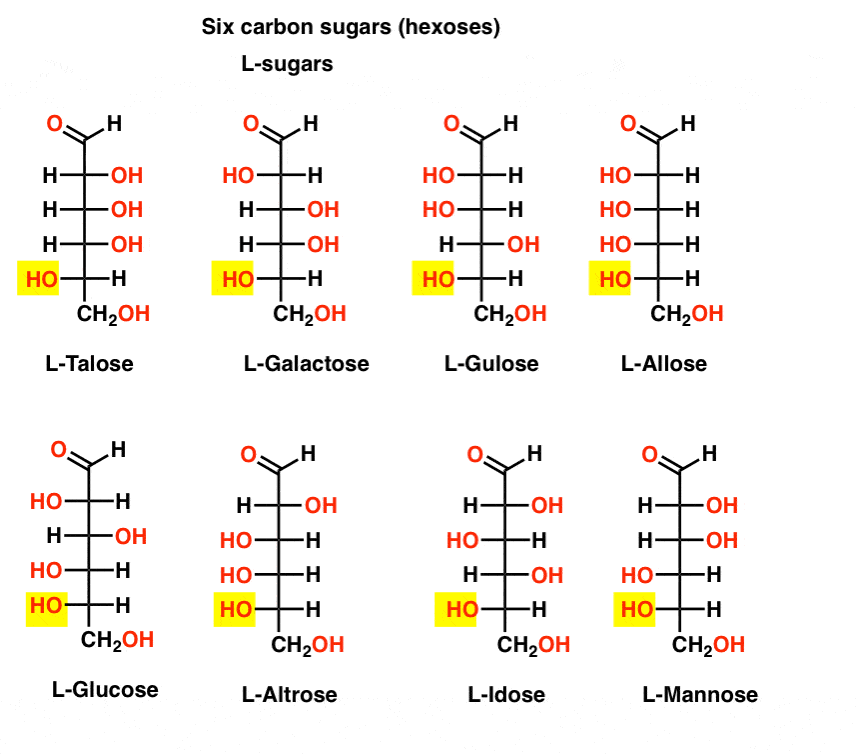
en contraste con los d-azúcares, los l – azúcares (abajo) rara vez se encuentran en la naturaleza. Curiosamente, la L-glucosa ha sido explorada como un sustituto del azúcar. Su sabor es indistinguible de la d-glucosa natural, pero no proporciona ningún alimento ya que no puede ser degradado por nuestras enzimas (quirales). Resulta que la producción es demasiado cara para competir con splenda, stevia, aspartame et. al.
OK, eso es suficiente. Se han producido azúcares de siete carbonos (aldoheptosis), pero no son biológicamente significativos.,
7. Pero espera, ¡hay más! (Aminoácidos)
si dibuja aminoácidos en la proyección de Fischer con el grupo más oxidado en la parte superior (el ácido carboxílico), también puede asignar L – Y D-.
de los 19 aminoácidos quirales que se incorporan a las proteínas (proteinogénico es el término apropiado) son todos L- . (La glicina es aquiral, por lo que D-y L-no se aplica). Algunos d-aminoácidos ocurren naturalmente, pero son raros (principalmente encontrados en bacterias, con la notable excepción del veneno de ornitorrinco) y no están codificados por ARNm.,
curiosamente, aunque los 19 aminoácidos quirales son L -, solo 18 de los 19 Son (S). ¿Cuál es la excepción?
( Esta es una buena trivia de la barra de Química Orgánica).
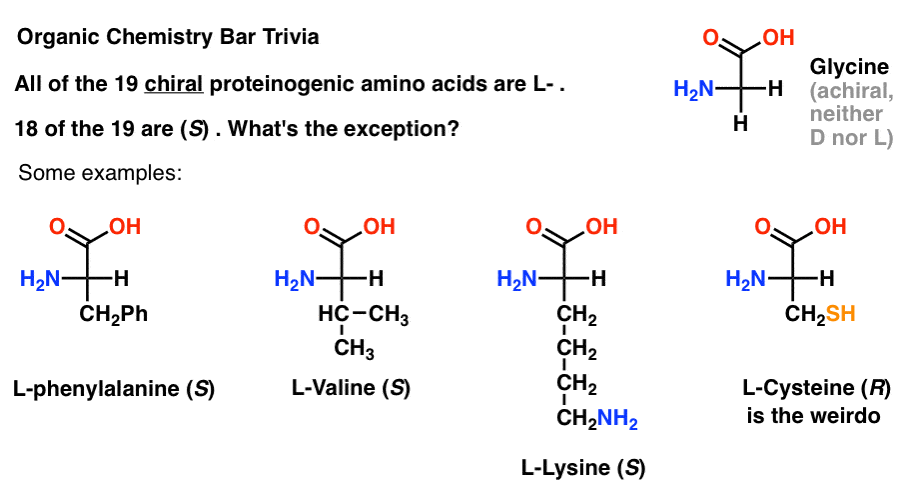
la Cisteína es el bicho raro.
(puntos de bonificación si también dijiste selenocisteína NER nerd).
resumen: Notación D – y L-para azúcares y aminoácidos
así que ese es el sistema D – L – para asignación de configuración absoluta. Funciona bien para los azúcares, ya que pueden acumularse de manera tan sistemática (como el sistema binario). También es útil para los aminoácidos., El punto clave es mirar el estereocentro inferior mientras se dibuja en la proyección de Fischer. ¿Verdad? D. ¿Izquierda? L.
Por supuesto, los azúcares no siempre se dibujan de manera tan útil en las proyecciones de Fischer: forman anillos. Vamos a escribir acerca de la determinación de D – y L-en azúcares cíclicos en un futuro post.
todo lo que tengo que decir es, agradecer a cualquier deidad que elijas creer en que la suposición de Fischer resultó ser correcta. Sería un enorme dolor en el culo para tamizar a través de 70+ años de la literatura química sabiendo que la configuración equivocada se había asignado a todos los azúcares y aminoácidos.,
Gracias a Thomas Struble por su ayuda con este post.
notas
Nota 1. «el penúltimo estereocentro en la mayoría de los azúcares quirales es R, mientras que el estereocentro en la mayoría de los aminoácidos es S «no tiene el mismo anillo, especialmente porque la cisteína es R.]
Nota. He aquí un experimento mental para determinar las configuraciones relativas de erythrose y threose., (Digo «experimento mental» porque no quiero incluir reactivos específicos, que podrían distraer)
si uno comienza con (+)-eritrosa pura y oxida el alcohol primario a un aldehído usando métodos conocidos, uno obtiene un compuesto que carece de cualquier rotación óptica. Lo mismo es cierto para (–)-erythrose, que devuelve un compuesto completamente idéntico. De esto se puede deducir que la estructura del nuevo compuesto debe ser tal que la molécula tenga un plano interno de espejo (es decir, es meso). .,
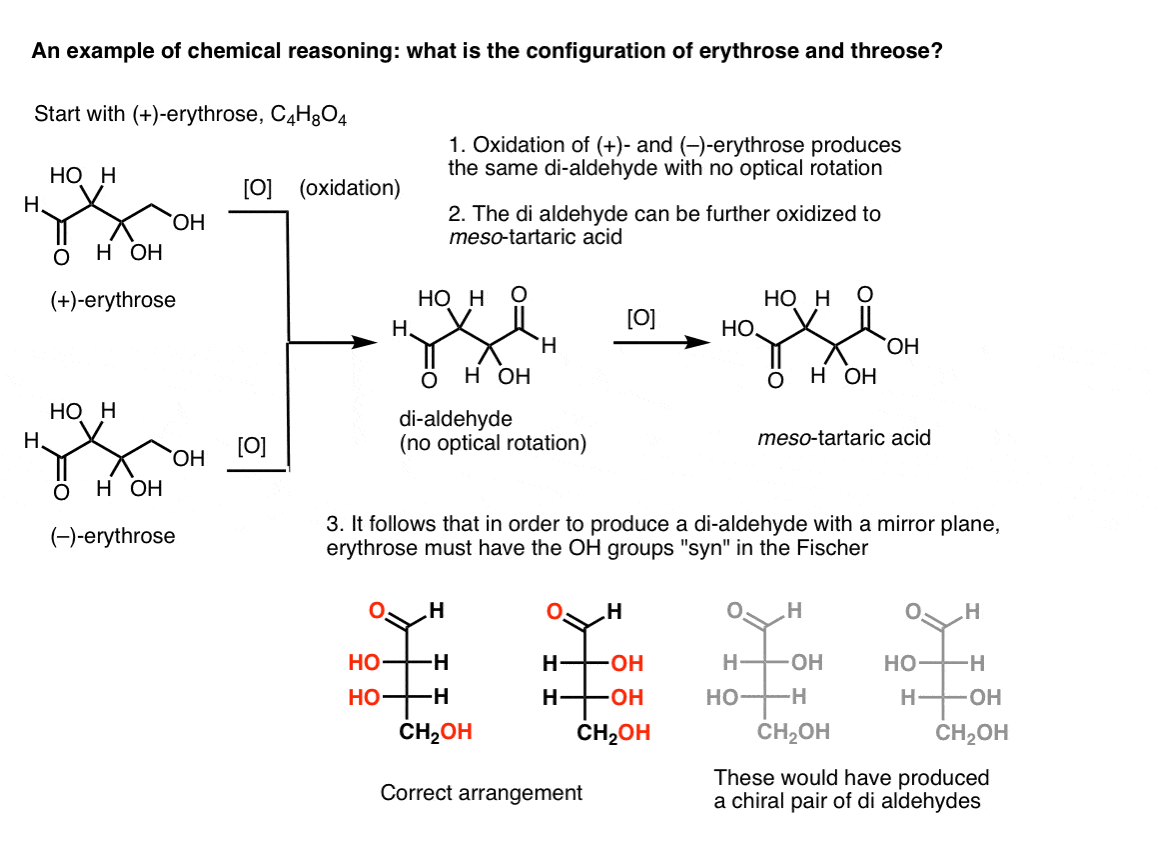
en contraste, realizar la misma operación en ( + )-threose resulta en un di-aldehído que mantiene la rotación óptica. Un compuesto con rotación óptica igual y opuesta se forma realizando la misma operación en (–)-threose. Estos dos compuestos son enantiómeros. Para que eso sea cierto, la orientación relativa de los grupos hidroxilo en treosa debe ser anti. Estos compuestos pueden oxidarse aún más para producir, respectivamente, ácido (–)- y (+)- tartárico.,
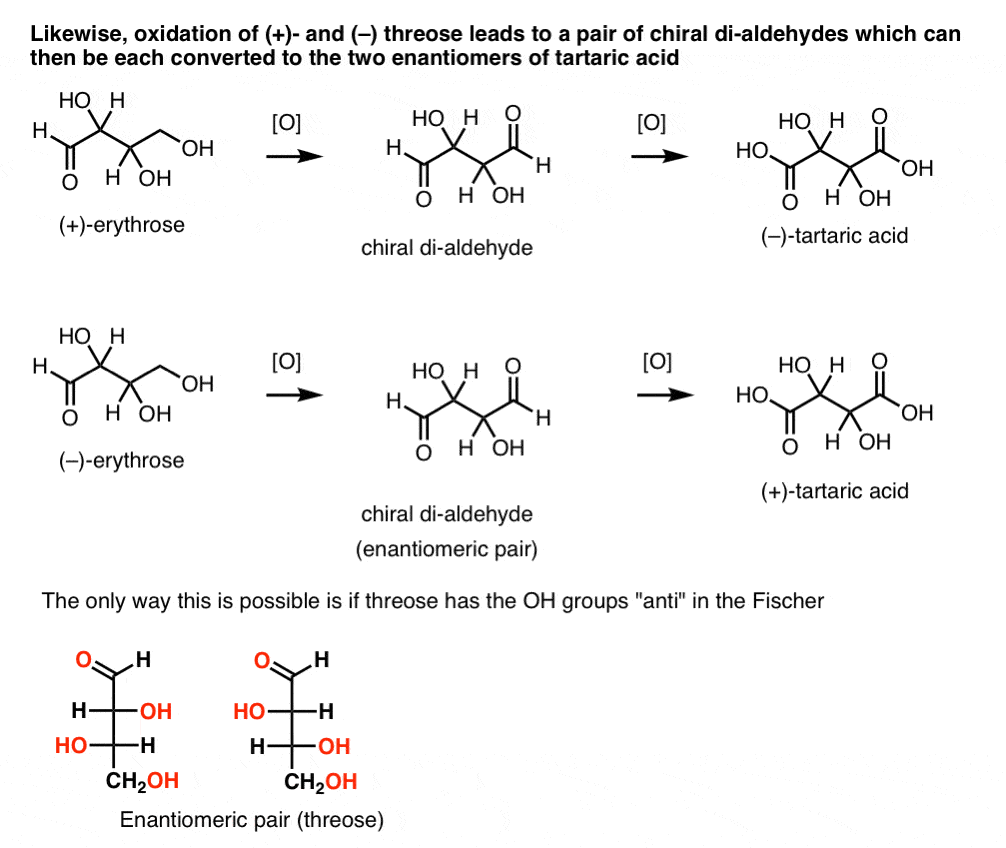
el mismo razonamiento se puede utilizar en la dirección opuesta (reducción). Por ejemplo ( + ) – o ( – ) – erythrose se puede reducir al eritritol tetra-ol, que es meso.
del mismo modo, la reducción de (+)- y (–) treosa resulta en un par enantiomérico de Tetra-ols, threitol.
de estos hechos podemos deducir la orientación relativa de los grupos OH.
(Advanced) References and Further Reading
- Über die Bezeichnung von optischen Antipoden durch die Buchstaben D und L
Emil Fischer
Ber. 1907, 40 (1), 102-106 DOI: 10.,1002 / cber.19070400111
Este es el famoso papel donde Prof. Emil Fischer (arbitrarily!) asignado ( – ) – gliceraldehído la L-estereoquímica. - Syntheses in the Purine and Sugar Group
Emil Fischer
Nobel Lecture, 1902Fischer Nobel Lecture, donde habla de su trabajo no solo en carbohidratos, sino también en purinas, de las cuales compuestos como la cafeína y la teobromina son miembros. El Prof. Fisher predice el aumento de las bebidas energéticas (p. ej., Red Bull), afirmando ,» con el ejercicio de un poco de imaginación se puede prever el día en que los granos ya no serán necesarios para hacer un buen café: una pequeña cantidad de polvo de un producto químico funciona junto con el agua proporcionará una bebida sabrosa y refrescante sorprendentemente barata».
- determinación de la configuración absoluta de compuestos ópticamente activos mediante rayos X
BIJVOET, J., PEERDEMAN, A. & van Bommel, A.,Nature 1951, 168, 271-272 DOI: 1038/168271a0 el famoso artículo que probó, usando el análisis estructural de rayos X, que «la Convención de Emil Fischer, que asignó la configuración de la Fig. 2 al ácido dextrorotatorio, parece responder a la realidad». - El descubrimiento de Emil Fischer de la configuración de la glucosa. A semicentennial retrospect C. S. Hudson Journal of Chemical Education 1941, 18 (8), 353 DOI: 10.1021/ed018p353 una revisión temprana que cubre el trabajo del Prof. Emil Fischer en química de carbohidratos.,
- La prueba de Emil Fischer de la configuración de los azúcares:un homenaje Centenario
Frieder W. Lichtenthaler
Angew. Chem. Int. Eréctil. 1992, 31 (12), 1541-1556 DOI: 10.1002/anie.199215413
Una revisión muy legible de 1992 que cubre el trabajo del Prof. Fischer en química de carbohidratos y profundiza en las asignaciones estereoquímicas de carbohidratos.