
A D – E L-notação para a configuração absoluta de açúcares e aminoácidos
O que diferencia “D-Glucose” de “L-Glucose” ? Ou d-alanine de L-alanine?
E o que é esta nomenclatura D E L, afinal? Leia em:
tabela de conteúdo
- D-E L-fornece uma abreviatura rápida para designar enantiômeros
- Por Que Nos preocupamos com esta nomenclatura antiga?,
- L – E D – Sistema De Atribuição de “Configuração Absoluta”
- Quatro-Carbono Aldeído D – e L – Açúcares (Aldotetroses)
- Cinco Carbono Aldeído D – e L – Açúcares (Aldopentoses)
- Seis Carbono Aldeído D – e L – Açúcares (Aldohexoses)
- Mas Espere, Há Mais! (Aminoácidos)
- Resumo: D – e L – Notação Para Açúcares e Aminoácidos
- Notas
- (Avançado) Referências e leituras Adicionais
D – E L – Fornece Um Rápido atalho Para Designar os Enantiómeros
D – e L – notação fornece um rápido atalho para a designação de enantiómeros.,
D-Glucose é o enantiómero da L-Glucose, por exemplo. Como L-Alanina é o enantiômero de D-alanina.
é atribuído como se segue. Para o açúcar, redigido em projeção de Fischer com mais oxidada do carbono no topo (i.e. aldeído ou cetona)
- se o OH no fundo centro quiral aponta para a direita, ele é conhecido como D-
- se o OH no fundo centro quiral aponta para a esquerda, ele é conhecido como L- .
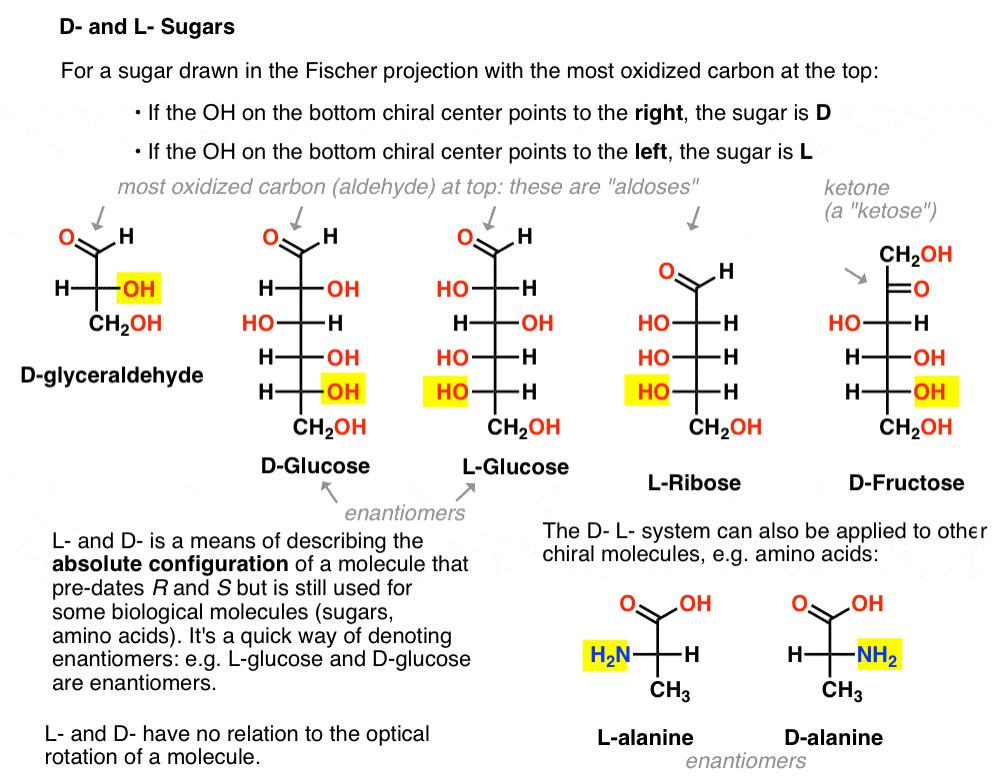
esta terminologia também pode ser aplicada aos aminoácidos: ver L – E D – alanina na figura acima.,2. Porque Nos Preocupamos Com Esta Nomenclatura Antiga?
você pode perguntar justificadamente: nós já não temos um sistema para atribuir configuração absoluta ? Para que precisamos de um novo sistema?o sistema D-L não é um novo sistema, pessoal. É o sistema antigo. é anterior ao Cahn-Ingold-Prelog.
o sistema D-L é literalmente um remanescente da era cavalo – e-buggy, que remonta ao trabalho de Emil Fischer sobre carboidratos no final de 1800-um tempo em que os químicos orgânicos não tinham como determinar a configuração absoluta dos estereocentros, que só se tornou possível em 1951 (thx, Bijvoet).,então porque é que ainda se usa? Não devia ser remetido para o caixote do lixo da história, juntamente com regras de deslizamento, cassetes de 8 faixas e disquetes de 5 ¼ de”?bem, há comunidades prósperas em partes da América rural onde as carruagens puxadas a cavalo persistem-se souber onde procurar. (Talvez algum dia haverá comunas onde as pessoas só usam tecnologia de computadores das décadas de 1970 e 1980?)
da mesma forma, há uma bolsa de química orgânica onde o sistema D-L ainda encontra uso, e isso é especificamente no Reino dos açúcares e aminoácidos.,esta não é uma revolta dos químicos Amish contra os males modernos do sistema CIP, a propósito. Existem pelo menos 3 boas razões, no caso específico dos açúcares e aminoácidos, para a utilização de L – E D- :
- brevidade. D-glucose é muito mais rápido para escrever e dizer que (2R,3S,4R,5R)2,3,4,5,6-pentahidroxihexanal. O sistema L – / D permite que a configuração de uma molécula com múltiplos centros quirais seja resumida com uma única letra (mais seu nome comum, é claro – graças a Noel para o lembrete)
- mais brevidade., Acontece que é uma maneira rápida de se referir aos enantiômeros. O enantiómero de L-glucose é D-glucose. O enantiómero de L-triptofano é D-triptofano. E enquanto nós poderíamos usar os prefixos ( + ) – ou ( – ) – para diferenciar os dois enantiômeros da glicose e outros açúcares, o sinal de rotação óptica pode variar com solvente, temperatura, concentração, e outros fatores que o tornam menos do que ideal. Além disso, L – E D – referem-se especificamente à configuração absoluta, enquanto (como vimos anteriormente) não existe uma relação simples entre o sinal de rotação óptica e configuração.,
- acontece que a maioria dos açúcares naturais são D -, e a maioria dos aminoácidos naturais são L -. Há uma enorme quantidade de informação comprimida nessa afirmação, e não há nenhum sistema concorrente (R/S,+/–) que poderia substituir o L – E D – por um único caráter. Nota
apresenta repetições: com açúcares e aminoácidos, L – E D – podem ser designações úteis. Para outras moléculas, você pode esquecer isso.
então o que é este sistema D-/L, e como esses termos se relacionam com a estrutura?,
Juntar-me à medida que viajam através do tempo…
A L – E D – Sistema De Atribuição de “Configuração Absoluta”
Emil Fischer começou a estudar hidratos de carbono no final da década de 1880. Ele era conhecido por esse tempo (através de van’t Hoff) que o carbono foi tetraédrica, e ele também era conhecido que moléculas contendo um carbono com quatro substituintes diferentes poderia girar-plano de luz polarizada (e.g. Pasteur). O que não se sabia era a configuração absoluta de qualquer uma das moléculas quirais – o que nos referiríamos hoje como suas configurações “R” E “S”.,
o hidrato de carbono mais simples contendo um centro quiral é o gliceraldeído, C3H6O3. Gliceraldeído tem três carbonos, tornando – se uma”triose”. O carbono mais oxidado no gliceraldeído é um aldeído, que também o torna um “aldose”.

em 1888, os dois enantiómeros do gliceraldeído foram isolados e caracterizados., Mas uma vez que não há uma correlação simples entre a configuração de um centro quiral e a direção na qual ele gira a luz polarizada plano, Emil Fischer não tinha nenhuma maneira de ligar de volta a rotação óptica de ( + )-e ( – ) –gliceraldeído para a configuração absoluta dos átomos em torno do centro quiral.usando a terminologia de hoje, ele não tinha como saber se ( – ) – gliceraldeído era (R) ou (s).
faltando esta peça chave de informação, Fischer escolheu adivinhar.,
A adivinhar, que acabou por ser correto, foi que o (–)-glyceraldehyde tinha a configuração do que hoje chamamos de S, e que (+)-glyceraldehyde tem a configuração que hoje chamamos de R.
claro, Cahn, Ingold e Prelog não tinha nascido ainda, e o sistema CIP só iria ser desenvolvido após Bijvoet do trabalho, em 1951. Então o Fischer desenvolveu a sua própria nomenclatura.
desenhando gliceraldeído no que mais tarde seria chamado de projeção de Fischer, ele atribuiu a configuração à esquerda para ( -) – gliceraldeído, e chamou-o de L – (abreviação de laevo Latino)., Ele então atribuiu a configuração no direito de (+)- gliceraldeído, e chamou – o de D – (para dextro Latino ).
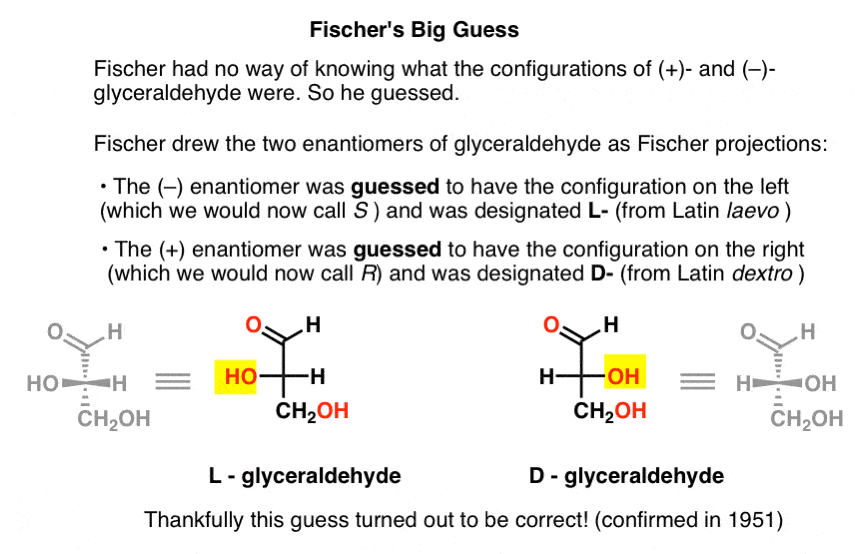
Por que isso é tão importante?atribuir a configuração absoluta para L-E D-gliceraldeído foi um pouco como atribuir o Meridiano de Prime (longitude 0°) ao Observatório Real em Greenwich, Inglaterra., Assim como a longitude de qualquer outro lugar na terra poderia então ser determinada em relação a esse ponto se suas distâncias relativas fossem conhecidas, a configuração absoluta de qualquer outro estereocentro poderia então ser determinada se sua configuração relativa a L – ou d – gliceraldeído fosse conhecida.
pode não ser a analogia mais clara. Então vamos olhar para os 4-açúcares de carbono para outro exemplo.,uma vez que as configurações absolutas de L – E D – gliceraldeído foram propostas, as configurações absolutas de outros compostos quirais poderiam então ser estabelecidas por analogia (e um monte de trabalho de grunt químico).
não é crucial para hoje, mas para um exemplo deste tipo de raciocínio, veja isto .
de volta quando o conceito de centros quirais estava sendo introduzido, você provavelmente aprendeu que uma molécula com centros quirais n terá estereoisômeros 2n (desde que não haja compostos meso).,existem duas aldoses de quatro carbonos, treose e eritrose. Cada um deles tem dois centros quirais. Cada um existe como um par de enantiômeros (L – E D- ) dando quatro estereoisômeros no total.aqui estão eles. A coisa importante a notar na figura abaixo é que a família L de açúcares tem o grupo OH do carbono quiral inferior à esquerda, e a família D tem o grupo OH do carbono quiral inferior à direita (destacado).,
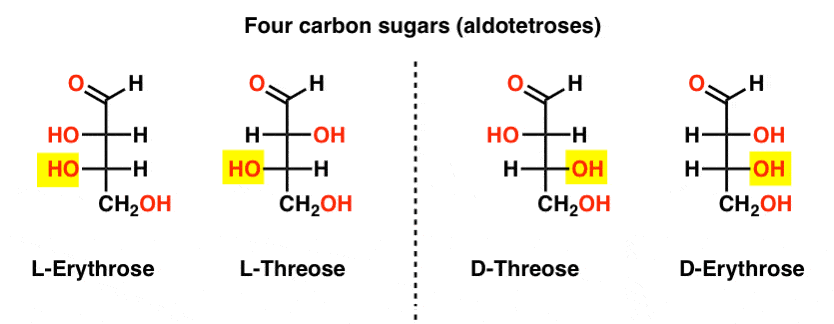
Veja como L-Erythrose e L-Threose construir no stereocenter estabelecido em L-glyceraldehyde (em destaque), e D-Erythrose e D-Threose construir no stereocenter estabelecido no D-glyceraldehyde (realçado).
açúcares são construídos um pouco como o sistema binário; você pode pensar que cada estereocentro é um “bit” que pode ter um de dois valores. A configuração de l-erythrose e L-threose difere apenas em um estereocentro. Se mudássemos a posição de H E oh, apanhávamos a outra., Esta relação tem um nome que você pode ver às vezes: duas moléculas que têm a configuração oposta em apenas um estereocentro são chamadas epimers, particularmente quando um dos átomos ligados ao estereocentro é um hidrogênio (H).
Cinco aldeídos de carbono D-E L-açúcares (Aldopentoses)
o nome mais familiar nessa lista deve ser ribose, que é a espinha dorsal de açúcar do ácido ribonucleico (ARN).
no lado esquerdo do diagrama abaixo, temos as l-aldopentoses, que todas compartilham a mesma configuração do estereocentro inferior quando o aldeído é colocado no topo.,
seus enantiômeros, as d-aldopentoses, estão do lado direito, que todos compartilham a mesma configuração do estereocentro inferior (realçado).
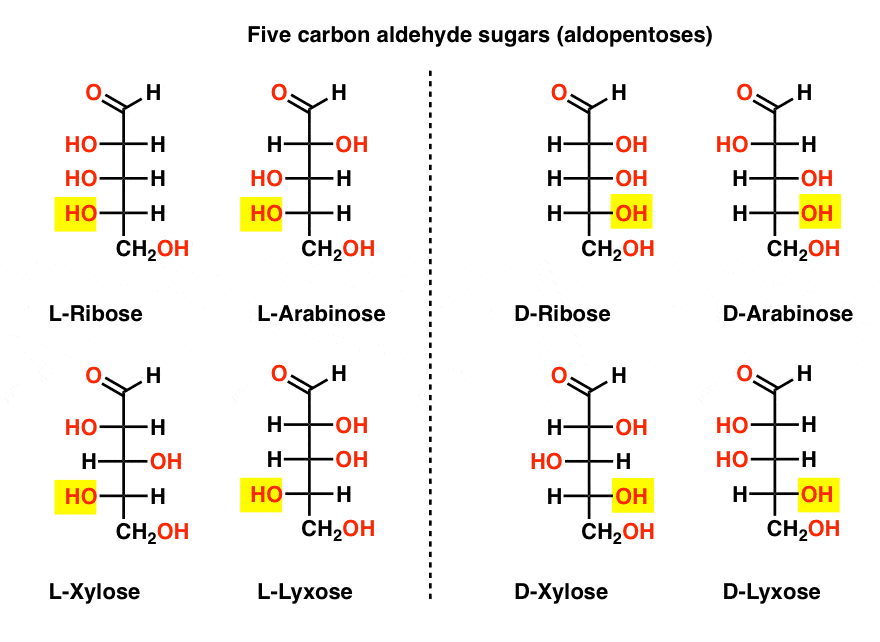
neste ponto, devemos salientar que a esmagadora maioria dos açúcares nas formas de vida terrestres são d-açúcares, incluindo D – ribose como a espinha dorsal do ARN. Por que e como todos os organismos na terra acabaram com D-Sugar é um mistério, como se presume que l-Sugar teria funcionado tão bem., se existem 4 aldopentoses, cada uma como um par D-L de enantiómeros (total de 8 estereoisómeros), então quantas Aldo-hexoses existem?
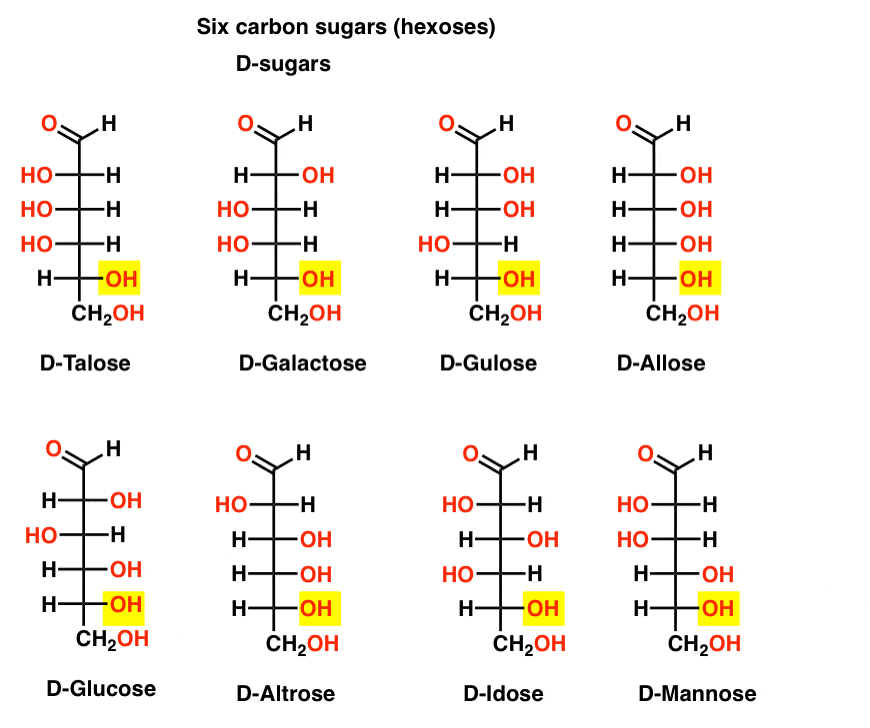
Existem 8 pares D-L (16 estereoisómeros no total). O mais familiar é a glucose, mas provavelmente reconhecerá manose e galactose. Alguns raramente, se alguma vez, são encontrados na natureza (idose, alguém?).
Aqui estão as D-Aldo-hexoses. Note como todos eles têm a mesma configuração do centro quiral inferior-o mesmo que vimos em D-gliceraldeído.,
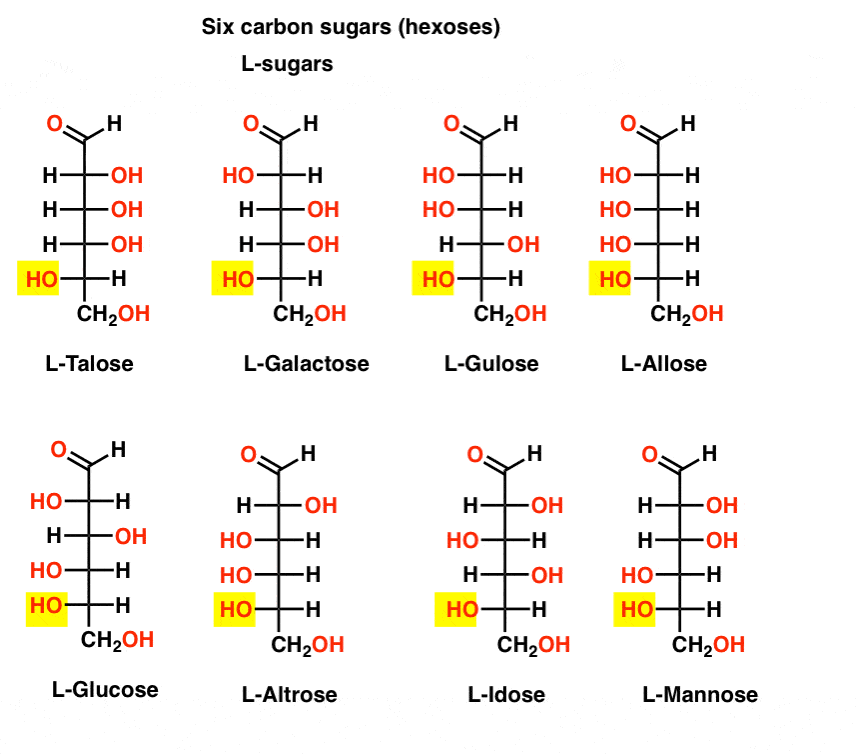
em contraste com os açúcares D, os açúcares L (abaixo) são raramente encontrados na natureza. Curiosamente, L-glucose tem sido explorado como um substituto do açúcar. O seu sabor é indistinguível da D-glucose natural, mas não fornece alimento, uma vez que não pode ser decomposto pelas nossas enzimas (quirais). Ao que parece, a produção é muito cara para competir com splenda, stevia, aspartame et. al.
OK, já chega. Os açúcares de sete carbonos foram feitos (aldoheptoses), mas não são biologicamente significativos.,
7. Mas espera-há mais! (Aminoácidos)
Se você desenhar aminoácidos na projeção Fischer com o grupo mais oxidado no topo (o ácido carboxílico), então você também pode atribuir L – E D-.
dos 19 aminoácidos quirais incorporados em proteínas (o termo proteinogénico é o correcto) são todos L- . (A glicina é achiral, por isso A D-E A L-não se aplica). Alguns D-aminoácidos ocorrem naturalmente, mas são raros (principalmente encontrados em bactérias, com a notável exceção do veneno de platypus) e não são codificados pelo ARNm.,curiosamente , embora todos os 19 aminoácidos quirais sejam L -, apenas 18 dos 19 São (S). Qual é a excepção?
(isto é um bom trivial de Química Orgânica).
cisteína é a estranha.
(Pontos bônus se você também disse selenocisteine … nerd).
resumo: Notação D – E L – para açúcares e aminoácidos
de modo que é o sistema D – L para atribuição da configuração absoluta. Funciona bem para os açúcares, uma vez que eles podem ser construídos de forma tão sistemática (como o sistema binário). Também é útil para aminoácidos., O ponto chave é apenas olhar para o estereocentro inferior enquanto ele é desenhado na projeção de Fischer. Certo? O D. Foi-Se Embora? L.
naturalmente, os açúcares nem sempre são tão úteis em projeções Fischer-eles formam anéis. Vamos escrever sobre a determinação de açúcares cíclicos em D – E L – in num futuro post.
tudo o que tenho a dizer é, obrigado a qualquer divindade que você escolher acreditar que o palpite de Fischer acabou por ser correto. Seria uma enorme dor de cabeça examinar mais de 70 anos da literatura química sabendo que a configuração errada tinha sido atribuída a todos os açúcares e aminoácidos.,graças a Thomas Struble pela ajuda neste post.
notas
Nota 1. “o penúltimo estereocentro na maioria dos açúcares quirais é R, enquanto o estereocentro na maioria dos aminoácidos é S” não tem o mesmo anel, especialmente porque a cisteína é R. ]
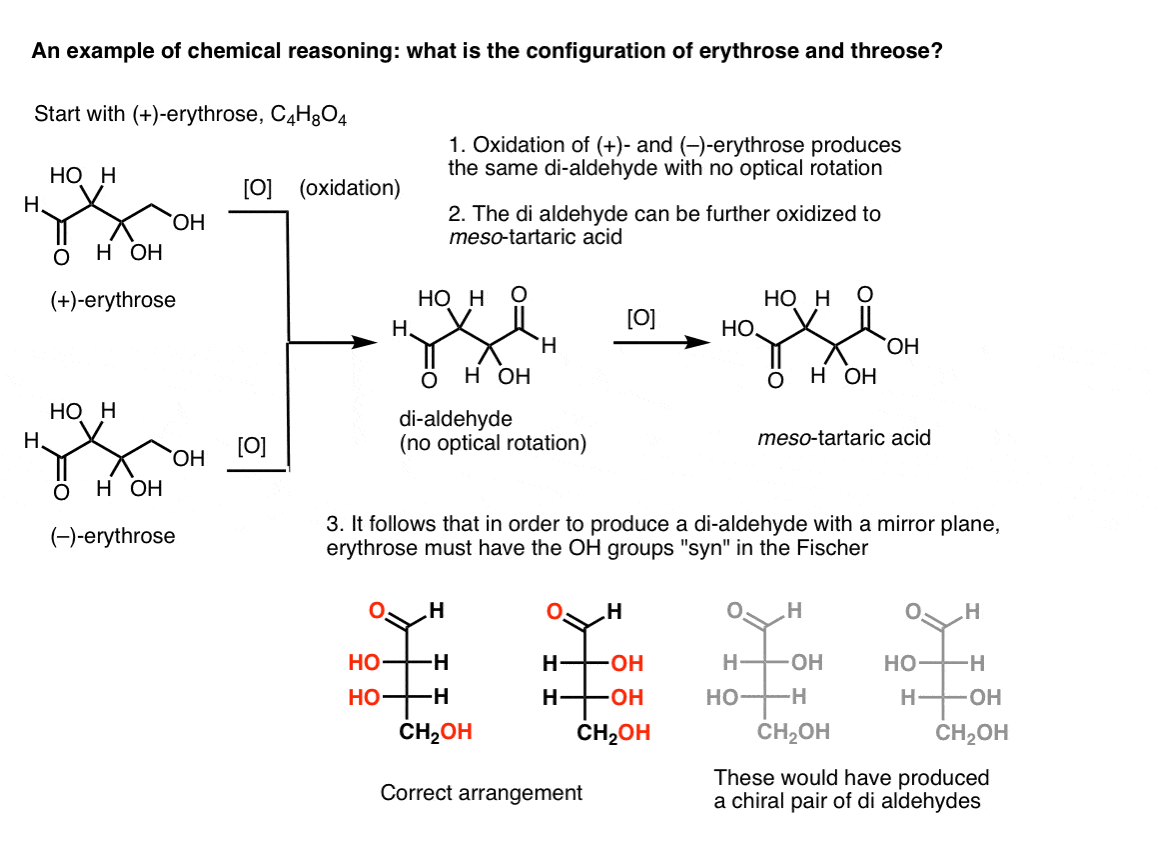
Nota. Aqui está uma experiência de pensamento para determinar as configurações relativas de erythrose e threose., (Eu digo “experiência de pensamento”, porque eu não quero incluem reagentes específicos, o que poderia ser um factor de distracção)
Se uma pessoa começa com puro (+)-erythrose e oxida o álcool primário para um aldeído usando métodos conhecidos, obtém-se um composto sem qualquer rotação óptica. O mesmo é verdadeiro para (–)-eritrose, que retorna um composto completamente idêntico. A partir disso, pode-se deduzir que a estrutura do novo composto deve ser tal que a molécula tem um plano de espelho interno (ou seja, é meso). .,
Em contraste, realizando a mesma operação em (+)-treose resulta em um di-aldeído que mantém rotação óptica. Um composto com rotação óptica igual e oposta é formado pela realização da mesma operação em ( – ) – treose. Estes dois compostos são enantiômeros. Para que isso seja verdade, a orientação relativa dos grupos hidroxilo em treose deve ser anti. Estes compostos podem ser oxidados para produzir, respectivamente, ácido (–)- e (+)- tartárico.,
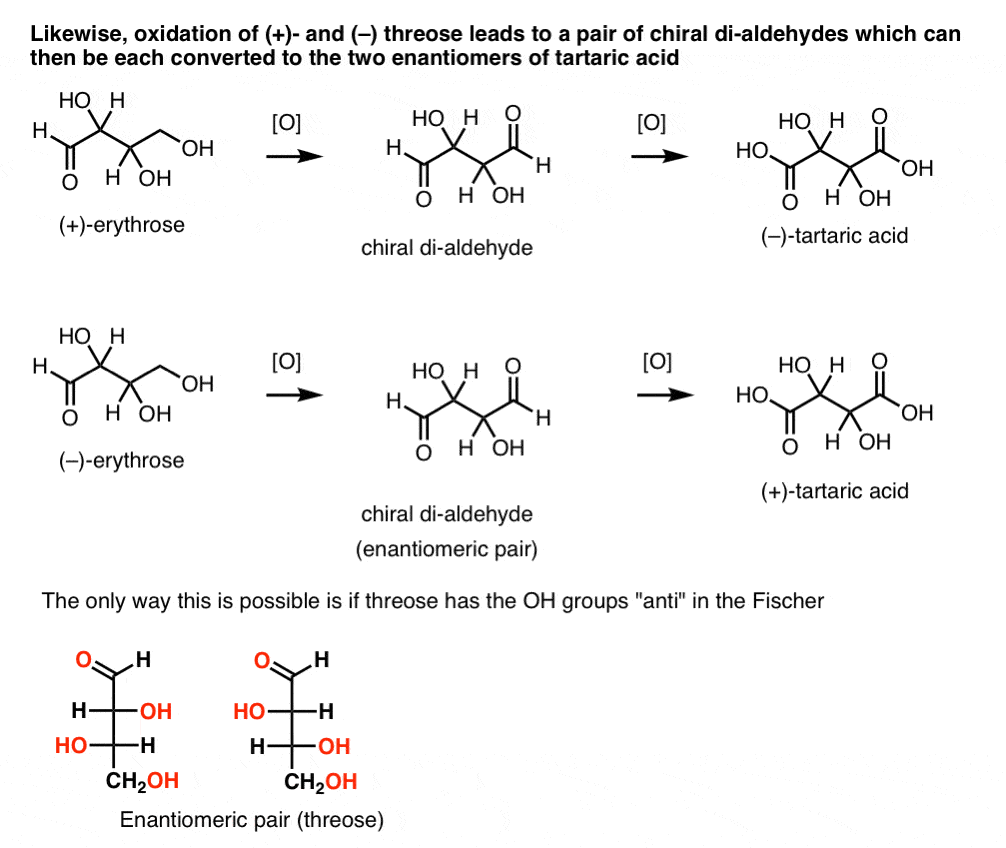
o mesmo raciocínio pode ser usado na direção oposta (redução). Por exemplo, a ( + ) – ou ( – ) – eritrose pode ser reduzida ao Tetra-ol eritritol, que é meso.da mesma forma, a redução de (+)- e (–) treose resulta em um par enantiomérico de tetra-ols, treitol.a partir destes fatos podemos deduzir a orientação relativa dos grupos OH.
(Advanced) References and Further Reading
- Über die Bezeichnung von optichen Antipoden durch die Buchstaben D und L
Emil Fischer
Ber. 1907, 40 (1), 102-106
DOI: 10.,1002 / RCE.19070400111
Este é o famoso artigo onde o Prof. Emil Fischer (arbitrariamente!) atribuiu (-) – gliceraldeído a l-estereoquímica. - Sínteses em Purinas e Açúcar do Grupo
Emil Fischer
a Palestra do Nobel, 1902Fischer Nobel da Palestra, onde ele fala sobre o seu trabalho e não apenas em hidratos de carbono, mas também em purinas, de que compostos como cafeína e teobromina são membros. O professor Fisher prevê o aumento das bebidas energéticas (ex., Red Bull), afirmando, “com o exercício de um pouco de imaginação o dia pode ser previsto quando os grãos não serão mais necessários para fazer um bom café: uma pequena quantidade de pó de um produto químico funciona juntamente com a água irá fornecer uma bebida saborosa, refrescante surpreendentemente barato”.Determination of the Absolute Configuration of Optically Active Compounds by Means of X-Rays BIJVOET, J., PEERDEMAN, A. div id= “2fbebf48b1”> van BOMMEL, A.,
Nature 1951, 168, 271-272
DOI: 1038 / 168271a0
The famous paper that proved, using X-ray structural analysis, that “Emil Fischer’s convention, which assigned the configuration of Fig. 2 to the dextrorotatory acid, appears to answer to reality”. - Emil Fischer descobriu a configuração da glicose. A semicentennial retrospect
C. S. Hudson
Journal of Chemical Education 1941, 18( 8), 353
DOI: 10.1021/ed018p353
An early review that covers Prof. Emil Fischer’s work in carbydrates chemistry., - Emil Fischer’s Proof of the Configuration of sugar: A Centennial Tribute
Frieder W. Lichtenthaler
Angew. Chem. T. Disfuncao. 1992, 31 (12), 1541-1556
DOI: 10.1002/anie.199215413
a very readable review from 1992 that covers Prof. Fischer’s work in carboidratos chemistry and goes in-depth into the stereochemical assignments of carboidratos.